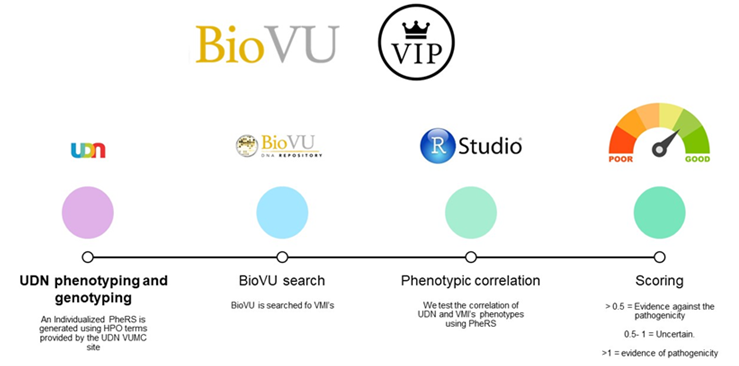
Personalized Structural Biology
Using software tools, our structural biology analyses estimate whether a genetic change, or variant, may affected a protein in multiple ways. This includes assessment of whether this may cause a disruption to protein folding, analysis of the proximity to other variants that are known to affect protein function, and prediction of the involvement of variants in protein interaction or modification. In addition to structure-based analyses, the pipeline runs a digenic predictor that estimates if interactions between genes to estimate whether variants in multiple genes may add together to contribute to disease. This also prepares 3D visualizations of sequence conservation. As the final automated step, the pipeline creates a patient case website of detailed calculation outputs and interactive protein visualizations.
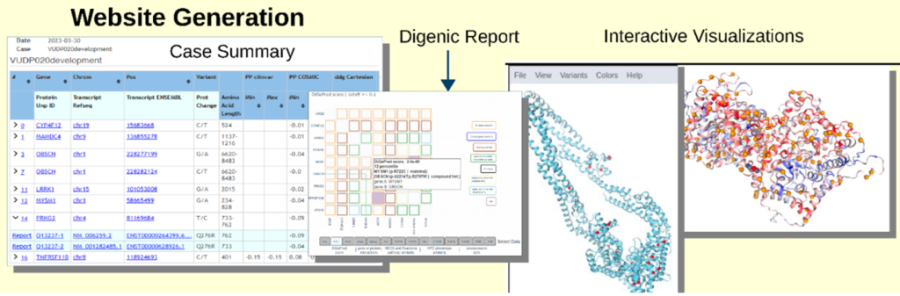
Predixcan
Predixcan or transcriptome wide association studies (TWAS) methods try to associate gene expression levels with phenotypes. By using a large expression data set coupled with genome sequencing, such as GTEx, we can identify single nucleotide polymorphisms (SNPs) that affect gene expression called eQTL (expression quantitative trait loci). These eQTLs can then be used to predict expression in samples where no expression data is available, such as BioVU. We use a machine-learning algorithm to select the SNPs used to predict the genetic component of gene expression. We can then test for associations between predicted gene expression and phenotypes in BioVU. Therefore, we can test candidate genes that have variants for association between predicted gene expression levels and phenotypes present in the proband. Because individuals with rare genetic disorders often contain loss of function variants, we can preferentially look for gene-phenotype associations where lower gene expression leads to the phenotype.
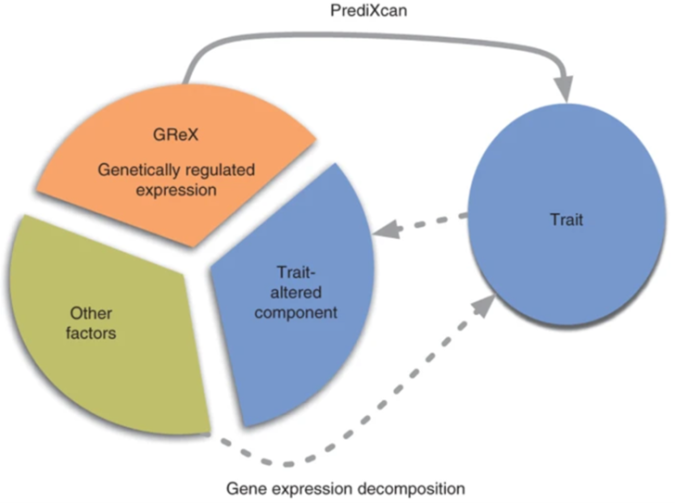
BioVU
The biomedical informatics team assesses rare genetic variants found in the genomes of undiagnosed patients. We have developed a preliminary pipeline that uses dense phenotype biobank data to generate a value of phenotype/genotype correlation for candidate variants found in undiagnosed patients. Our pipeline uses BioVU, a de-identified biobank developed and housed at VUMC that links electronic health records (EHRs) to genotypic data. The BioVU variant interpretation pipeline (BioVU VIP) is a fully automated program that analyzes candidate variants selected by the UDN clinical team. Using the clinical description, it builds an EHR-compatible phenotype profile known as a phenotypic risk score (PheRS) that is applied to a cohort of 30,000 BioVU patients. It then scans the available genotype data for individuals with the same candidate variant as the individual. Finally, BioVU VIP uses regression analysis to test the hypothesis that VMIs are phenotypically similar to the individual, comparing the PheRS of BioVU individuals with the candidate variant and those without. We produce a report of candidate variants for presentation to the clinical team at the weekly case analysis meeting.